
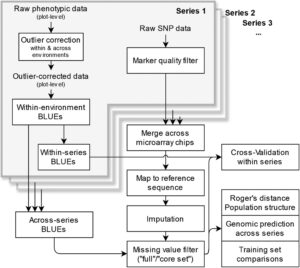
Ringkasan
Big data, dikombinasikan dengan teknik kecerdasan buatan (AI), memiliki potensi untuk meningkatkan akurasi prediksi genom secara signifikan. Termotivasi oleh keberhasilan yang dilaporkan untuk hibrida gandum, kami memperluas cakupan ke galur murni dengan mengintegrasikan data fenotipik dan genotipik dari empat program pemuliaan gandum komersial. Bertindak sebagai wali amanat data akademis, kami menggabungkan data ini dengan seri eksperimen historis dari kemitraan publik-swasta sebelumnya. Data terintegrasi mencakup 12 tahun, 168 lingkungan, dan menyediakan set pelatihan prediksi genom hingga ~9500 genotipe untuk hasil gabah, tinggi tanaman, dan tanggal pembuahan. Meskipun data fenotipik dan genotipik heterogen, kami dapat memperoleh data berkualitas tinggi dengan menerapkan kurasi data yang ketat, termasuk imputasi SNP. Kami menggunakan data untuk membandingkan prediksi genomik linier terbaik yang tidak bias dengan prediksi genomik berbasis jaringan saraf konvolusional. Analisis kami mengungkapkan bahwa kami dapat menggabungkan rangkaian eksperimen secara fleksibel untuk prediksi genomik, dengan kemampuan prediksi yang terus meningkat seiring bertambahnya ukuran set pelatihan, mencapai puncaknya pada sekitar 4000 genotipe. Seiring bertambahnya ukuran set pelatihan, perolehan kemampuan prediksi menurun, mendekati titik jenuh jauh di bawah batas teoritis yang ditentukan oleh akar kuadrat heritabilitas. Jalan potensial, seperti set pelatihan yang dirancang atau pendekatan prediksi non-linier baru, dapat mengatasi titik jenuh ini dan membantu memanfaatkan big data bernilai tinggi yang dihasilkan dengan lebih maksimal dengan memecah silo data di seluruh perusahaan.
Perkenalan
Dalam dekade terakhir, prediksi genom secara luas telah merevolusi pemuliaan tanaman dengan memberikan estimasi nilai genotipe varietas kandidat baru dari profil genomik dan pengamatan fenotipe genotipe terkait (Meuwissen et al ., 2001 ). Hal ini memungkinkan beberapa uji lapangan yang mahal untuk dihilangkan, karena tahap uji pemuliaan awal dengan heritabilitas rendah yang diamati dapat digantikan menggunakan prediksi genomik (Riedelsheimer dan Melchinger, 2013 ). Dengan demikian, waktu yang diperlukan untuk memilih genotipe unggul dapat dipersingkat (Beyene et al ., 2021 ), bahkan untuk sifat kompleks yang dikendalikan oleh banyak gen.
Mengingat banyaknya jumlah gen dan interaksinya yang memengaruhi sebagian besar sifat agronomi, ukuran populasi yang layak dalam program pemuliaan tidak memungkinkan kita untuk menyimpulkan pengaruh setiap lokus dan interaksi lokus individu. Teknik statistik modern telah dikembangkan untuk mengatasi kekurangan model linear tradisional dalam menangkap interaksi gen yang kompleks dan hubungan genotipe, yang secara efektif mengurangi pengaruh sejumlah besar data yang bising (Chafai et al ., 2023 ). Metode yang paling praktis dan banyak digunakan adalah model parametrik atau semi-parametrik (Montesinos-López et al ., 2022 ). Model-model ini umumnya memperkenalkan asumsi apriori tentang efek genetik, baik dengan regularisasi estimasi parameter (de los Campos et al ., 2013 ) atau dengan memilih distribusi prior informatif dalam kerangka Bayesian (Gianola, 2013 ). Awalnya, parameter yang akan diestimasi adalah efek lokus genetik, seperti dalam Ridge-Regression Best Linear Unbiased Prediction (rrBLUP, Meuwissen et al ., 2001 ), tetapi efek genetik individu dapat dimodelkan secara langsung dengan memasukkan korelasi yang diharapkan dari nilai pembiakan mereka dalam model. Ketika silsilah tidak diketahui, hubungan ini dapat disimpulkan dari data genomik, yang memunculkan Genomic BLUP (GBLUP, VanRaden, 2008 ). Matriks kekerabatan genomik yang dihasilkan mempertimbangkan hubungan aditif-genetik, yaitu, silsilah individu, dan kelompok hubungan bersama, yang menghubungkan lokus Single Nucleotide Polymorphism (SNP) yang diuji ke lokus kausatif (Habier et al ., 2013 ). Akibatnya, populasi yang lebih beragam mungkin menjadi target yang lebih sulit untuk prediksi genomik, karena kelompok hubungan yang lebih banyak dan lebih kecil harus diperhitungkan (Daetwyler et al ., 2013 ).
Dalam beberapa tahun terakhir, pendekatan pembelajaran mendalam untuk prediksi genomik telah mendapatkan perhatian (Ma et al ., 2018 ; Montesinos-Lopez et al ., 2021 ). Berbeda dengan metode konvensional yang disebutkan di atas, metode ini tidak menggunakan model genetik kuantitatif tetapi didasarkan pada pengaturan fleksibel dari banyak transformasi non-linier dari data masukan (neuron) untuk mendeteksi (1) pola dalam data masukan, dan (2) hubungannya dengan fenotipe. Parameter transformasi tersebut dioptimalkan oleh pembelajaran terbimbing pada set pengujian. Hal ini diharapkan memberikan keuntungan ketika sifat tanaman sangat dipengaruhi oleh efek interaksi kompleks yang tidak tercakup oleh teori di balik salah satu model yang lebih klasik (Pérez-Enciso dan Zingaretti, 2019 ). Selain itu, pelatihan jaringan saraf memiliki kompleksitas waktu linier sehubungan dengan ukuran sampel. Hal ini menghindari ledakan waktu komputasi yang dihadapi peneliti setiap kali matriks kekerabatan harus dibalik, seperti dalam kasus GBLUP (Pook et al ., 2020 ). Sementara pada awalnya, multilayer perceptrons telah digunakan untuk prediksi genomik, baru-baru ini jaringan konvolusional telah menunjukkan kemampuannya untuk menangkap pola keterkaitan (Pook et al ., 2020 ). Namun, potensi penuh dari pembelajaran mendalam belum sepenuhnya terwujud dengan menyediakan set pelatihan yang jauh lebih besar daripada yang tersedia untuk satu lembaga. Dengan demikian, evaluasi komprehensif tentang kelebihan dan keterbatasan teknik pembelajaran mendalam ketika diterapkan pada set data besar sangat dibutuhkan, khususnya dalam domain pemuliaan tanaman.
Kemampuan prediksi prediksi genomik, yang didefinisikan sebagai korelasi antara fenotipe sebenarnya dan yang diprediksi, dipengaruhi secara signifikan oleh beberapa karakteristik set pelatihan dan pengujian, seperti (1) ukuran populasi, (2) keragaman dan keterkaitan antara genotipe, (3) tingkat ketidakseimbangan hubungan (LD) dan (4) kualitas data fenotipe dan genotipe (Schopp et al ., 2016 ). Meningkatkan set pelatihan adalah strategi yang mudah dan menjanjikan untuk mencapai tingkat kemampuan prediksi yang tinggi untuk populasi yang beragam dalam praktik pemuliaan gandum hibrida (Zhao et al ., 2021 ). Beberapa langkah ke arah ini telah diambil. Misalnya, dalam sebuah penelitian yang mencakup lebih dari 8000 ras gandum lokal, kemampuan prediksi 0,68 untuk berat seribu biji dalam populasi dapat dicapai (Crossa et al ., 2016 ). Dalam studi lain, kumpulan data besar yang berisi lebih dari 10.000 galur gandum difenotipe dalam desain satu tahun yang tidak direplikasi, dan kemampuan prediksi yang mendekati satu untuk hasil gabah dapat dicapai dalam validasi silang (Norman et al ., 2018 ). Hasil ini sangat menggembirakan tetapi memerlukan investasi sumber daya yang besar di luar jangkauan sebagian besar lembaga dan perusahaan atau intensitas fenotipe yang dikurangi yang berada di bawah standar pemuliaan gandum komersial untuk pengembangan varietas dalam hal jumlah lingkungan dan replikasi.
Kolaborasi berbagai lembaga dan/atau lintas perusahaan untuk keuntungan bersama merupakan konsep yang menarik untuk meningkatkan populasi untuk melatih model prediksi genom secara luas tetapi terhambat oleh data yang heterogen dan non-ortogonal (tidak seimbang). Uji coba pemuliaan komersial tidak seimbang karena mereka menyaring sejumlah besar genotipe dan mengevaluasi fenotipe mereka hanya dalam sejumlah kecil lingkungan. Genotipe terpilih dari tahap pemuliaan pertama kemudian dievaluasi di lebih banyak lingkungan di musim berikutnya. Oleh karena itu, ketika keandalan estimasi kinerja kandidat meningkat, jumlah kandidat yang tersedia berkurang. Menggabungkan beberapa uji coba tersebut akan menghasilkan kumpulan data yang mencakup sejumlah besar genotipe tahap awal, tetapi juga data tahap akhir untuk sejumlah besar kandidat daripada yang layak untuk setiap aktor individu. Dalam studi sebelumnya, yang kami bangun di sini, menggabungkan beberapa uji coba gandum historis menggandakan kemampuan prediksi untuk hasil gabah untuk hibrida (Zhao et al ., 2021 ). Oleh karena itu, menggabungkan kumpulan data yang berbeda menjanjikan. Dalam studi ini, kami menyelidiki dampak pada prediksi genomik dari penggabungan data historis tersebut dengan data pembiakan rutin dari empat perusahaan. Tujuan kami adalah (1) untuk menyelidiki apakah mungkin untuk melakukan analisis terpadu dari kumpulan data fenotipik dan genotipik yang berbeda dan bagaimana melakukan kontrol kualitas dari tugas tersebut, (2) untuk memeriksa kemampuan prediksi apa yang dapat diharapkan ketika menggunakan prediksi genomik di luar batasan seri eksperimen individual dan seberapa baik beberapa seri dapat digabungkan untuk membentuk set pelatihan yang lebih besar untuk prediksi genomik serta untuk mengeksplorasi potensi model pembelajaran mendalam untuk meningkatkan proses ini dan (3) untuk menguji pendekatan untuk meningkatkan set pelatihan dengan mengambil subset dari data lengkap, menyaring data yang paling dapat diandalkan dan berpotensi meningkatkan kemampuan prediksi.
Hasil
Tidak adanya subpopulasi yang berbeda secara genetik yang terungkap melalui data genotipe yang diperhitungkan secara akurat
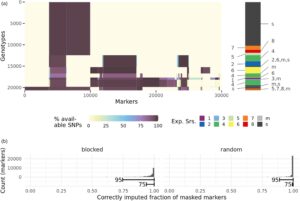
Mengingat kesenjangan blok-bijaksana dalam data SNP yang dihasilkan dari integrasi platform array SNP heterogen (Gambar 1 dan 2a ), kami melakukan validasi akurasi imputasi. Ini diperkirakan dengan menutupi dan mengimputasi beberapa data SNP dalam pendekatan yang diblokir dan acak dan kemudian menghitung rasio panggilan SNP yang diimputasi dengan benar terhadap panggilan SNP yang ditutupi. Dalam pendekatan yang diblokir, hampir semua penanda diimputasi dengan akurasi di atas 0,75 (persentil ke-95) dan sebagian besar (persentil ke-75) bahkan di atas 0,93 (Gambar 2b ). Imputasi menggunakan pendekatan masking acak dimungkinkan dengan akurasi yang lebih tinggi. Persentil 95% dan 75% dari akurasi imputasi masing-masing adalah 0,89 dan 0,99.
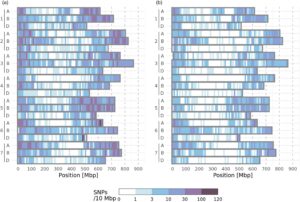
Data SNP yang diimputasikan mencakup sebagian besar genom gandum (Gambar 3a ). Penanda yang memenuhi kriteria nilai hilang liberal (maksimal 80% nilai hilang dan diimputasikan) ditemukan pada kepadatan sekitar 1–100 penanda per 10 Mbp (Gambar 3a ). Set penanda yang lebih kecil yang dihasilkan dari kriteria nilai hilang yang ketat, yaitu, dengan maksimal 30% nilai hilang dan diimputasikan, mencakup genom pada 1–30 penanda per Mbp (Gambar 3b ). Cakupan di dekat pusat kromosom jauh lebih lemah daripada di dekat ujung, dan terutama untuk kriteria nilai hilang yang ketat, celah besar terdapat di dekat pusat kromosom.
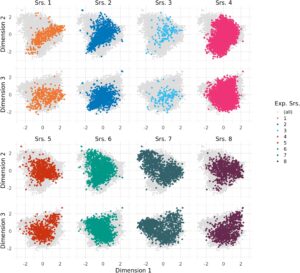
Analisis koordinat utama berdasarkan jarak Rogers mengungkap keragaman tambahan dengan menggabungkan rangkaian penelitian (Gambar 4 ). Bagian dari beberapa rangkaian ditemukan di wilayah ruang keragaman yang hanya sedikit dicakup oleh rangkaian lainnya. Misalnya, bagian dari rangkaian 6 dan 7 berada di luar ruang keragaman rangkaian 1–3 tetapi cukup mirip satu sama lain. Selain kecenderungan ke arah komplementaritas ini, tidak ada rangkaian yang membentuk klaster yang jelas berbeda dan terpisah dari semua rangkaian lainnya.
Data fenotipik memiliki kualitas tinggi dan konsisten dengan data genotipik
Kami telah mengintegrasikan dan menyusun data fenotipik yang dihasilkan dalam 105.000 petak hasil panen biji-bijian sebagai bagian dari kemitraan publik-swasta atau program pemuliaan gandum di Eropa Tengah. Sebagai ukuran kualitas data fenotipik, heritabilitas arti luas diperkirakan untuk tiga sifat untuk setiap seri eksperimen (Gambar 5a ). Heritabilitas berkisar dari 0,86 hingga hampir 0,98 diperoleh untuk tanggal pembuahan, dari 0,81 hingga 0,99 untuk tinggi tanaman, dan dari 0,74 hingga 0,93 untuk hasil panen. Dengan demikian, kualitas data fenotipik sangat baik untuk semua sifat.
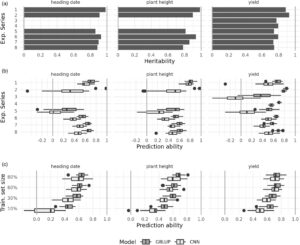
Validasi silang dalam seri menunjukkan kemampuan prediksi sedang hingga tinggi. Median di 20 replikasi berkisar dari 0,50 hingga 0,98, kecuali untuk seri 5 untuk tanggal pembuahan (0,26) dan seri 3 untuk hasil gabah (0,36, Gambar 5b ). Yang terakhir dapat dijelaskan oleh fakta bahwa seri 3 hanya terdiri dari 142 galur murni (Tabel 2 ). Untuk seri 5–8, yang mencakup data dari program pemuliaan gandum komersial, kemampuan prediksi untuk tanggal pembuahan dan tinggi tanaman lebih rendah daripada untuk seri historis. Ini dapat dijelaskan oleh sifat seri yang tidak seimbang ini. Karena kendala efisiensi ekonomi, sifat-sifat ini dievaluasi dalam lingkungan yang lebih sedikit daripada hasil gabah; dalam beberapa kasus, jumlah rata-rata lingkungan per genotipe bahkan
<2 (Tabel 1 ). Dalam keadaan ini, dampak pada kemampuan prediksi diharapkan. Secara keseluruhan, data SNP memberikan prediksi yang baik dari data fenotipik, yang menunjukkan integrasi data yang berhasil.
| Seri | Tahun (20) | Tanggal judul | Tinggi tanaman | Hasil panen gabah | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bahasa Inggris | rata-rata lingkungan | lingkungan total | kamu | Bahasa Inggris | rata-rata lingkungan | lingkungan total | kamu | Bahasa Inggris | rata-rata lingkungan | lingkungan total | kamu | ||
| 1 | 09–10 | 380 | 8.0 | 8 | 2 | 380 | 8.0 | 8 | 2 | 380 | 8.0 | 8 | 2 |
| 2 (jam) | 16–19 | 3639 | 17.6 | 38 | 4 | 3639 | 18.3 | 39 | 4 | 5051 | 10.1 | 61 | 4 |
| 2 (saya.) | 16–19 | 469 | 18.5 | 38 | 4 | 469 | 19.6 | 39 | 4 | tahun 1099 | 9.6 | 61 | 4 |
| 3 (jam) | 12–13 | tahun 1604 | 11.0 | 11 | 2 | ||||||||
| 3 (saya.) | 12–13 | 144 | 11.0 | 11 | 2 | ||||||||
| 4 | 12–15 | 4958 | 3.9 | 30 | 4 | ||||||||
| 5 | tahun 20–21 | 781 | 1.3 | 7 | 1 | tahun 1001 | 2.1 | 8 | 1 | Tahun 1911 | 4.4 | 26 | 2 |
| 6 | tahun 20–21 | tahun 1631 | 5.3 | 13 | 2 | tahun 1631 | 5.6 | 15 | 2 | tahun 1631 | 5.7 | 17 | 2 |
| 7 | tahun 20–21 | tahun 1707 | 4.1 | 9 | 2 | tahun 1742 | 4.0 | 10 | 2 | tahun 1742 | 4.4 | 12 | 2 |
| 8 | tahun 20–21 | 3516 | 1.4 | 7 | 2 | 3512 | 1.6 | 7 | 2 | 3505 | 2.1 | 15 | 2 |
| (semua) | Tanggal 09–21 | 12.096 tahun | 8.1 | 81 | 8 | 12 347 | 8.3 | 83 | 8 | 21.891 | 6.2 | 168 | 12 |
Jaringan saraf konvolusional menjadi kompetitif dengan ukuran set pelatihan yang lebih besar
Ukuran set pelatihan sangat memengaruhi kinerja yang dicapai dengan jaringan saraf konvolusional (CNN). Untuk ukuran set pelatihan kecil sebesar 10% (untuk hasil: sekitar 950 genotipe, Tabel 2 ), CNN menunjukkan kemampuan prediksi yang lebih rendah dengan margin sekitar 0,15 dibandingkan dengan GBLUP (selisih median, Gambar 5c ). Menariknya, saat ukuran set pelatihan meningkat menjadi 80% (sekitar 7.600 genotipe untuk hasil), margin ini menyempit dan CNN berkinerja serupa dengan GBLUP, bahkan melampauinya pada beberapa iterasi individual (Gambar 5c ). Tingkat penutupan celah ini bervariasi dengan sifat dan untuk hasil, di mana set pelatihan terbesar tersedia, celah ditutup lebih awal daripada yang lain.
| Seri | Ne | Jumlah genotipe | |||
|---|---|---|---|---|---|
| Tanggal judul | Tinggi tanaman | Hasil panen gabah | (semua) | ||
| 1 | 34.5 | 371 | 371 | 371 | 371 |
| 2 | 51.6 | 467 | 467 | tahun 1081 | tahun 1081 |
| 3 | 49.1 | angka 0 | angka 0 | 142 | 142 |
| 4 | 58.0 | angka 0 | angka 0 | 3703 | 3703 |
| 5 | 61.9 | 214 | 418 | 641 | 641 |
| 6 | 55.0 | tahun 1614 | tahun 1614 | tahun 1614 | tahun 1614 |
| 7 | 29.7 | tahun 1178 | tahun 1213 | tahun 1213 | tahun 1213 |
| 8 | 40.7 | 848 | 848 | 848 | 848 |
| (semua) | 79.4 | 4665 | 4904 | 9480 | 9480 |
Kami juga membandingkan kinerja CNN dengan GBLUP, dengan data yang disilo ke dalam seri eksperimen individual. Ukuran set pelatihan untuk prediksi ini adalah 90% dari hitungan genotipe seri eksperimen individual, yang merupakan 128 hingga 3332 genotipe dalam contoh yield (Tabel 2 ). Kemampuan prediksi GBLUP yang tinggi untuk seri dikaitkan dengan kinerja CNN yang kuat, hanya menyisakan sedikit perbedaan antara kemampuan prediksi CNN dan GBLUP (Gambar 5b ): Misalnya, untuk yield, seri 2 dan 4 menghasilkan prediksi GBLUP paling akurat (0,85 dan 0,69) dan juga gap terkecil (0,06 dan 0,13) antara prediksi GBLUP dan CNN. Gambarannya terbalik untuk seri 3, dengan median kemampuan prediksi GBLUP terburuk (0,36) dan gap terbesar terhadap median kemampuan prediksi CNN (0,46). Satu-satunya outlier pada pola ini adalah seri eksperimen 2 untuk tanggal heading dan tinggi tanaman, di mana kemampuan prediksi GBLUP yang tinggi bertepatan dengan kesenjangan yang besar pada kinerja CNN.
Kami tidak menyelidiki kinerja CNN pada sisa penelitian tetapi menggunakan GBLUP karena beban komputasi yang tinggi dan kinerja GBLUP yang sebanding atau lebih unggul.
Rangkaian eksperimen dapat digabungkan secara fleksibel dalam set pelatihan prediksi genomik, mendekati titik puncak dalam kemampuan prediksinya
Bahasa Indonesia: Untuk menguji kemampuan memprediksi genotipe yang tidak diketahui yang diberikan kumpulan seri dalam studi ini, kami memilih berbagai kombinasi seri sebagai set pelatihan dan menetapkan semua seri lainnya sebagai set uji. Kami kemudian memperoleh kemampuan prediksi genomik dengan membandingkan prediksi dengan BLUE lintas-seri dari set uji, menghitung kemampuan prediksi secara terpisah untuk setiap seri dalam set uji. Mengumpulkan berbagai kombinasi dan jumlah seri menghasilkan set pelatihan dengan ukuran berbeda, yang dengannya kemampuan prediksi diurutkan (Gambar 6a ). Sebagai tren umum, kami mengamati peningkatan kemampuan prediksi dengan peningkatan ukuran set pelatihan, tetapi peningkatan ini mendekati dataran tinggi di luar ukuran set pelatihan sekitar 4000 individu. Untuk banyak set uji, kemampuan prediksi mendekati yang diperoleh dengan menggunakan data seri itu sendiri dalam prediksi genomik yang divalidasi silang (Gambar 6a , garis putus-putus) tetapi tidak mendekati batas atas yang ditentukan oleh akar kuadrat heritabilitas (garis utuh).
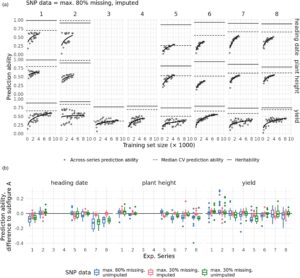
Skenario di atas didasarkan pada data penanda yang diimputasikan dan kriteria penyaringan SNP liberal (<80% dari nilai yang hilang sebelum diimputasikan). Konsisten dengan akurasi imputasi yang tinggi dari data SNP yang hilang, kami juga mengamati penurunan kemampuan prediksi dalam prediksi genom secara rata-rata di ketiga sifat ketika kriteria penyaringan liberal yang sama digunakan tetapi tidak ada nilai yang hilang yang diimputasikan, atau ketika kriteria penyaringan SNP yang ketat (<30% dari nilai yang hilang sebelum diimputasikan) digunakan (Gambar 6b ). Dengan demikian, penyaringan liberal yang dikombinasikan dengan imputasi juga tampaknya menjadi strategi yang paling berhasil untuk data penanda yang hilang per blok yang mendasari set data yang digunakan.
Kami kemudian menyelidiki apakah jumlah seri yang disertakan dalam set pelatihan memiliki efek signifikan pada kemampuan prediksi, mengingat ukuran set pelatihan yang tidak berubah. Kami membuat set pelatihan 800 genotipe yang berasal dari seri eksperimen tunggal atau jamak dan membandingkan kemampuan prediksi yang dihasilkan: Rata-rata, menggunakan seri tunggal menghasilkan kemampuan prediksi yang lebih rendah sebesar 0,02 untuk tanggal heading dan di bawah 0,01 untuk tinggi tanaman dan hasil gabah (Gambar S1 ). Deviasi standar antara replikasi jauh lebih besar, berkisar antara 0,07 hingga 0,08. Perbedaan deviasi standar di bawah ±0,01 untuk semua sifat. Jadi, untuk set pelatihan berukuran sedang, jumlah seri eksperimen yang disertakan dalam set pelatihan tidak memiliki efek yang berarti pada kemampuan prediksi.
Sebagian besar rangkaian percobaan kompatibel satu sama lain untuk prediksi
Dengan mengamati peningkatan kemampuan prediksi genomik dengan meningkatnya ukuran set pelatihan, kami menyelidiki apakah penyertaan seri individual dalam set pelatihan menguntungkan kemampuan prediksi untuk semua atau beberapa set pengujian. Sebagai titik awal untuk analisis ini, kami memperoleh peningkatan rata-rata kemampuan prediksi dengan ukuran set pelatihan menggunakan model empiris (Persamaan 5 , Gambar 6a ). Kami mempertimbangkan residual, yang dapat diartikan sebagai kinerja menjalankan prediksi genomik, dikoreksi untuk ukuran set pelatihan. Kami memperhatikan bahwa penyimpangan ini kecil dibandingkan dengan efek ukuran set pelatihan, dengan deviasi standar 0,09 untuk hasil gabah dan tanggal pembuahan, dan 0,05 untuk tinggi tanaman. Ini juga dapat dilihat secara visual, dengan kemampuan prediksi genomik berkumpul secara dekat di sekitar kecenderungan umum untuk sebagian besar set pengujian (Gambar 6a ).
Kami kemudian menguraikan penyimpangan dari prediksi genomik berjalan menjadi kontribusi seri individu dalam set pelatihan mereka ( 6 ) untuk mengetahui apakah seri eksperimen tertentu menyebabkan prediksi berjalan secara sistematis melebihi atau kurang berkinerja. Pengaruh pilihan seri yang dikoreksi untuk ukuran set pelatihan kecil dibandingkan dengan pentingnya meningkatkan ukuran set pelatihan (Gambar S2b ). Total varians yang diatribusikan ke seri individu atau kombinasinya lebih tinggi untuk tanggal heading (0,009) daripada untuk tinggi tanaman dan hasil gabah (0,003). Dilihat dari ukuran relatif komponen varians, efek interaksi dari pasangan seri eksperimen tertentu (satu menjadi bagian dari set pelatihan, yang lain menjadi set uji) adalah faktor yang mendominasi. Efek utama dari seri individu yang berada di set pelatihan, yang mewakili seri eksperimen yang meningkatkan atau menurunkan kemampuan prediksi untuk sebagian besar set uji, hanya memainkan peran kecil (Gambar S2b ). Hanya beberapa kombinasi set pelatihan—set uji individu yang luar biasa bermanfaat atau merugikan untuk kemampuan prediksi. Seri 1 dan 2 menunjukkan kompatibilitas yang sangat tinggi, meningkatkan kemampuan prediksi sebesar 0,1–0,3 di atas nilai yang diharapkan untuk ukuran set pelatihan untuk ketiga sifat yang dipelajari (Gambar S2a ). Selain itu, kombinasi dengan deviasi terkuat dari ekspektasi tidak menunjukkan pola tertentu (Gambar S2a ), misalnya, seri 5 → 7 (set pelatihan → set pengujian), dengan kemampuan prediksi sebesar -0,09 dibandingkan dengan ekspektasi, atau seri 7 → 8 dan sebaliknya untuk tanggal heading (+0,07/+0,09).
Memastikan keberagaman lingkungan meningkatkan kinerja set pelatihan
Untuk menguji strategi yang dapat lebih meningkatkan kinerja set pelatihan untuk prediksi genomik, kami memilih set pelatihan yang mencakup sejumlah lingkungan atau tahun yang ditentukan, sambil menjaga ukuran set pelatihan tetap konstan. Ukuran set pelatihan moderat (300 dan 600 untuk pendekatan pertama dan kedua, masing-masing) dihasilkan dari pembatasan dari sifat data yang tidak seimbang. Mengikuti strategi pertama, set pelatihan yang didukung oleh jumlah lingkungan yang lebih tinggi cenderung menghasilkan akurasi prediksi yang lebih baik (Gambar 7a ). Namun, dibandingkan dengan set pelatihan dengan ukuran yang sama yang diambil sampelnya dari set data lengkap tanpa pembatasan jumlah lingkungan, pilihan ini tidak atau hanya sedikit lebih baik. Untuk tanggal heading dan tinggi tanaman, set pelatihan yang didukung oleh setidaknya enam lingkungan per genotipe cukup untuk mencapai kemampuan prediksi set acak. Untuk hasil gabah, kemampuan prediksi set acak dicapai dengan setidaknya empat lingkungan per genotipe.
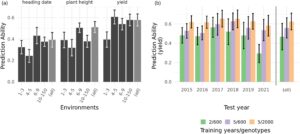
Jumlah tahun yang lebih banyak dalam set pelatihan dikaitkan dengan kemampuan prediksi yang lebih baik. Untuk semua tahun dalam data yang tersedia, memasukkan lima tahun sebelumnya dalam set pelatihan terbukti lebih unggul daripada memasukkan 2 tahun dalam set pelatihan pada ukuran set pelatihan konstan 600 genotipe (Gambar 7b ).
Diskusi
Interoperabilitas yang memadai untuk memastikan keberhasilan integrasi data genotipe dan fenotipik
Sebelumnya, telah ditunjukkan bahwa data genotipik dan fenotipik dari berbagai proyek kemitraan publik-swasta dapat berhasil diintegrasikan, menggandakan kemampuan prediksi kinerja hibrida dalam gandum (Zhao et al ., 2021 ). Dalam studi ini, kami memperluas upaya ini ke data dari galur murni yang dihasilkan dalam empat program pemuliaan komersial (seri 5–8) dan set data lain yang diterbitkan (seri 1, Gogna et al ., 2022 ). Pertanyaan pertama yang harus dijawab adalah apakah data genotipik dan fenotipik baru tersebut dapat dioperasikan bersama, yang dapat terhambat baik oleh perbedaan metode dan protokol yang digunakan maupun oleh alasan biologis.
Platform genotyping yang digunakan merupakan faktor signifikan yang memengaruhi interoperabilitas data genomik (Gogna et al ., 2022 ; Schulthess et al ., 2022a ). Dalam studi ini, semua seri eksperimen menggunakan array SNP, khususnya array Illumina 90 k iSelect (Wang et al ., 2014 ). Namun, platform genotyping lain yang digunakan dalam penelitian gandum mungkin memiliki tumpang tindih terbatas atau sebagian (Sun et al ., 2020 ) dan platform ini mungkin lebih cocok untuk mengkarakterisasi populasi dengan ketidakseimbangan hubungan yang lebih kecil atau substruktur genetik yang berbeda. Akibatnya, memastikan interoperabilitas data genotyping di seluruh platform mungkin memerlukan upaya tambahan. Lebih jauh lagi, ketersediaan urutan penanda sangat penting untuk imputasi data genomik dalam pendekatan kami, meskipun ada metode alternatif yang tidak bergantung pada urutan penanda, meskipun dengan pengorbanan dalam akurasi (He et al ., 2015 ). Teknologi Genotyping-by-Sequencing (GBS), yang tidak digunakan dalam penelitian ini, menghasilkan data genotipe dengan kepadatan tinggi dan menunjukkan pola acak dari nilai yang hilang. Kesenjangan ini dapat diperhitungkan dengan akurasi yang lebih tinggi dibandingkan dengan kesenjangan sistematis yang timbul dari penggunaan platform penanda yang berbeda, seperti yang ditunjukkan dalam penelitian ini dan penelitian sebelumnya (He et al ., 2015 ; Torkamaneh dan Belzile, 2015 ). Oleh karena itu, kami optimis tentang integrasi sumber data tersebut. Ketika bekerja dengan populasi ras asli yang beragam, di mana ketidakseimbangan hubungan sering berkurang dan/atau terdapat subpopulasi yang berbeda secara genetik (Schulthess et al ., 2022b ), pendekatan analitis khusus mungkin diperlukan untuk memastikan interoperabilitas data genotipe.
Interoperabilitas data fenotipik juga menghadirkan tantangan bagi integrasi data. Perlu ada kesepakatan tentang definisi sifat-sifat yang terekam. Dalam kasus kami, kami diuntungkan oleh fakta bahwa semua data difokuskan pada Jerman, dan dengan demikian metode yang digunakan dalam uji varietas resmi Jerman (Uji Nilai untuk Budidaya dan Penggunaan) berfungsi sebagai standar de facto evaluasi sifat. Demikian pula, untuk alasan yang sama, rezim pertanian dalam penelitian kami serupa, menggunakan praktik pengelolaan intensif untuk menilai potensi hasil gabah. Untuk melampaui cakupan ini, standar dokumentasi metode umum perlu disetujui, digunakan, dan ditingkatkan, dengan menyeimbangkan garis tipis antara mencakup banyak kasus penggunaan dan dipahami serta digunakan dengan benar oleh praktisi (Darnala et al ., 2023 ; Papoutsoglou et al ., 2020 ; Selby et al ., 2019 ). Meski begitu, fenotipe objektif dapat menjadi tugas yang menantang dalam program pemuliaan komersial dengan mempertimbangkan ukuran jaringan uji, kecepatan perkembangan tanaman, dan keterbatasan jumlah staf.
Bahasa Indonesia: Untuk memungkinkan interoperabilitas antar seri, genotipe umum sangat penting untuk memperkirakan dampak lingkungan. Seri eksperimen dari penelitian ini sebagian besar dihubungkan oleh lebih dari lima genotipe, yang merupakan standar yang juga digunakan dalam pemuliaan tanaman komersial. Beberapa kombinasi seri berada di bawah standar ini (Tabel S1 ), khususnya untuk seri historis dengan sifat yang mengepalai tanggal dan tinggi tanaman. Untungnya, konektivitas set data juga dapat bersifat sementara, sehingga satu seri eksperimen yang terhubung dengan baik dengan dua seri lainnya dapat berfungsi sebagai referensi umum, yang menghubungkannya satu sama lain bahkan ketika mereka saling terhubung dengan lemah. Mendeklarasikan genotipe dengan jarak Rogers di bawah ambang batas tertentu sebagai sama (Zhao et al ., 2021 ) dapat lebih jauh meningkatkan jumlah ini sedikit (rata-rata tiga individu dalam kasus kami) untuk perbandingan berpasangan.
Potensi dan keterbatasan prediksi genom secara menyeluruh di seluruh seri
Bahasa Indonesia: Untuk memprediksi performa gandum hibrida, diketahui bahwa prediksi lintas populasi diuntungkan dengan menggabungkan beberapa seri eksperimen berbeda menjadi satu set pelatihan (Zhao et al ., 2021 ): Kemampuan prediksi hingga 0,4 tercapai. Di sini, kami fokus pada galur murni dan secara drastis meningkatkan jumlah genotipe yang tersedia dengan bekerja sama dengan pemulia komersial. Kami juga mengonfirmasi untuk galur murni bahwa prediksi lintas populasi diuntungkan dengan menggabungkan beberapa seri eksperimen berbeda menjadi satu set pelatihan dan sangat terkejut dengan fleksibilitas di mana seri eksperimen berbeda dapat digabungkan. Peningkatan kemampuan prediksi sebagian besar ditentukan oleh ukuran set pelatihan (Gambar 6a ), tetapi seri mana yang disertakan dalam set pelatihan kurang penting (Gambar S2b ). Faktor penting yang berkontribusi terhadap hal ini adalah struktur populasi yang lemah dalam populasi gabungan kami (Gambar 4 ). Studi lain, khususnya yang berfokus pada materi bank gen seperti ras asli, telah menemukan populasi yang membentuk klaster yang lebih jelas (Crossa et al ., 2016 ; Ramstein dan Casler, 2019 ; Schulthess et al ., 2022a ). Karena keterkaitan antara genotipe dalam set pengujian dan pelatihan menurun, kemampuan prediksi GBLUP juga menurun (Alemu et al ., 2023 ; Habier et al ., 2010 ; Lorenz dan Smith, 2015 ). Oleh karena itu, ketika populasi ditemukan berbeda dalam keragaman genetiknya, kekuatan prediksi lintas populasi akan menurun. Namun, kelompok pemuliaan gandum elit Eropa Tengah tampaknya berbagi sebagian besar keragaman genetik. Kami menyimpulkan bahwa dalam populasi tersebut, menggabungkan data dari beberapa program pemuliaan merupakan strategi yang sangat menjanjikan untuk meningkatkan kemampuan prediksi genomik dan menunjukkan cara bagi program pemuliaan skala kecil hingga menengah untuk mencapai manfaat bersama melalui kerja sama. Rangkaian eksperimen 2 sebagian berisi sumber daya genetik dari bank gen IPK, tetapi PCoA (Gambar 4 ) tidak menunjukkan ruang genetik yang jauh lebih besar yang dicakup oleh rangkaian ini dibandingkan dengan rangkaian lainnya. Alasannya bisa jadi karena keragaman ini, yang dibobot dengan jumlah genotipe, kurang menonjol dibandingkan dengan jarak antara set data individual dan dengan demikian tidak ditunjukkan oleh PCoA. Alasan lainnya bisa jadi karena penyaringan penanda sebelum jarak Roger dihitung, yang mungkin telah menghilangkan alel langka yang berkontribusi pada keragaman populasi tersebut. Namun, kemampuan prediksi dalam dan lintas tidak lebih rendah untuk rangkaian ini (Gambar 5b dan 6a)), yang menunjukkan bahwa tidak ada pengaruh kuat dari penanda yang dihilangkan pada sifat-sifat yang diukur. Sifat-sifat seperti ketahanan terhadap penyakit, yang dapat menunjukkan ketergantungan yang lebih tinggi pada lokus individu, akan memerlukan pendekatan yang lebih cermat dalam hal ini.
Peluang untuk lebih meningkatkan kemampuan prediksi
Memperluas set pelatihan di luar 4000 baris menghasilkan pengembalian yang semakin berkurang dalam kemampuan prediksi, dengan kinerja mendekati dataran tinggi. Namun, perlu dicatat bahwa dataran tinggi ini tercapai jauh di bawah heritabilitas set pengujian, yang mewakili maksimum teoritis (Gambar 6a ). Alasan untuk ini belum jelas dan memerlukan studi lebih lanjut. Pertama, ketika berpindah dari prediksi dalam seri ke lintas seri, seseorang kehilangan kemampuan prediksi tambahan yang diberikan GBLUP dengan menangkap ko-segregasi ketika genotipe pelatihan dan pengujian hanya berjarak beberapa generasi (Habier et al ., 2013 ). Kemungkinan penyebab lainnya adalah bahwa meningkatnya keragaman data gabungan melemahkan hubungan antara lokus penyebab dan lokus dalam data SNP (Meuwissen, 2009 ). Dalam studi sebelumnya, untuk mengukur efek ini dalam pengaturan terapan, subset dengan ukuran populasi nominal dan efektif yang berbeda diambil dari seri eksperimen 4 dan akurasi prediksi yang divalidasi silang, yaitu, kemampuan prediksi dibagi dengan akar kuadrat heritabilitas, diperoleh (Zhao et al ., 2021 ). Dengan mengekstrapolasi hubungan ini ke ukuran populasi nominal dan efektif yang terlihat dalam studi kami untuk populasi gabungan (Tabel 2 ), akurasi prediksi yang diharapkan sekitar 0,75 akan dihasilkan. Ini akan menjelaskan sebagian besar akurasi prediksi yang hilang dari set pelatihan dalam studi ini (Gambar S3 ). Menurut teori ini, meningkatkan rasio ukuran populasi nominal terhadap efektif akan meningkatkan kemampuan prediksi. Ini dapat dilakukan dengan memilih subset untuk data pelatihan lengkap yang sangat terkait dengan populasi uji. Namun, upaya sebelumnya untuk melakukan ini jarang berhasil melampaui kemampuan prediksi GBLUP menggunakan data pelatihan lengkap dan sebagian besar berfokus pada pencapaian daya yang sebanding dengan ukuran set pelatihan yang lebih kecil (Fernández-González et al ., 2024 ; Isidro y. Sánchez dan Akdemir, 2021 ; Lopez-Cruz dan de los Campos, 2021 ).
Jalan lebih jauh mungkin untuk menguji metode prediksi genomik alternatif; misalnya, BayesB dilaporkan berkinerja sedikit lebih baik daripada GBLUP dalam memperkirakan efek penanda berdasarkan LD ke lokus kausatif (Habier et al ., 2010 ). Baru-baru ini, upaya untuk menggunakan metode pembelajaran mesin non-linier, seperti Random Forest, Support Vector Machine atau Neural Networks untuk memprediksi kinerja genotipe, telah mencapai paritas dengan GBLUP dan terkadang bahkan mencapai hasil yang lebih baik (Abdollahi-Arpanahi et al ., 2020 ; Montesinos-López et al ., 2024 ; Sandhu et al ., 2021 ). Untuk menghubungkan dengan temuan ini, kami telah menggunakan Convolutional Neural Network (CNN) untuk prediksi genomik dan membandingkan hasilnya dengan GBLUP. Kami menemukan bahwa untuk tugas kami, CNN mampu memprediksi fenotipe sama baiknya dengan GBLUP, di mana GBLUP memberikan kemampuan prediksi yang tinggi. Akurasi prediksi GBLUP yang di bawah standar dikaitkan dengan kinerja CNN yang tidak hanya lebih lemah dalam istilah absolut tetapi juga menunjukkan kesenjangan kinerja yang lebih lebar dengan GBLUP. Tampaknya beberapa faktor yang merugikan GBLUP memengaruhi CNN secara tidak proporsional lebih kuat. Faktor yang paling mungkin adalah ukuran set pelatihan. Untuk prediksi lintas-seri dengan set pelatihan kecil (Gambar 5c ) dan prediksi dalam-seri dari seri eksperimen kecil (Gambar 5b ), kinerja CNN yang buruk diharapkan. Untuk menyesuaikan CNN, sejumlah besar parameter dan hiper-parameter harus diestimasi. Sebaliknya, untuk GBLUP, hanya parameter mean dan varians yang harus diestimasi, dan prediksi kemudian dapat diturunkan melalui teori genetik kuantitatif. Oleh karena itu, set pelatihan kecil diharapkan tidak cukup untuk pelatihan CNN yang memadai, dan akibatnya, GBLUP tetap menjadi model yang disukai untuk ukuran set pelatihan yang lebih kecil dari sekitar 4.000 genotipe. Pengecualian menarik untuk tren ini adalah rangkaian eksperimen 1, di mana CNN dan GBLUP mencapai akurasi prediksi tinggi meskipun ukuran set pelatihannya kecil.
Kinerja prediksi yang sebanding yang diamati dari GBLUP dan CNN konsisten dengan contoh dari literatur (Montesinos-López et al ., 2024 ; Sandhu et al ., 2021 ). Namun, penelitian tersebut hanya menggunakan sebagian kecil genotipe yang tersedia untuk penelitian ini. Oleh karena itu, masuk akal bahwa mungkin ada lebih banyak faktor daripada ukuran sampel yang menghambat kemampuan prediksi yang lebih baik dan keunggulan akhir prediksi berbasis jaringan saraf atas metode linier seperti GBLUP. Menariknya, mungkin ada padanan untuk jaringan saraf untuk hipotesis yang disebutkan di atas bahwa peran hubungan aditif-genetik yang semakin berkurang dalam set data multi-asal dapat menghambat prediksi GBLUP. Jaringan saraf dapat dengan kuat mendasarkan prediksinya pada hubungan aditif-genetik, mengabaikan efek dan interaksi alel individu. Fenomena ‘pembelajaran jalan pintas’ ini diamati sebelumnya untuk prediksi genomik dengan jaringan saraf (Ubbens et al ., 2021 ). Jaringan saraf memiliki banyak properti yang sangat menjanjikan untuk prediksi genomik, seperti kompleksitas waktu linier dengan peningkatan ukuran sampel, fleksibilitasnya untuk menggabungkan banyak tipe data yang berbeda seperti kovariat lingkungan (Washburn et al ., 2021 ), dan fleksibilitasnya dalam kasus di mana model genetik aditif gagal menggambarkan arsitektur genetik (Pérez-Enciso dan Zingaretti, 2019 ). Semakin banyak data pelatihan yang dapat dioperasikan dapat disediakan, mungkin melalui kerja sama lintas batas kelembagaan, semakin banyak potensi ini dapat direalisasikan. Dalam studi ini, IPK telah berfungsi sebagai ‘wali amanat data akademis’. Dengan menyediakan penyimpanan data yang netral dan rahasia, pemangku kepentingan komersial dapat menyumbangkan data. Hasilnya menunjukkan bahwa model seperti itu dapat memfasilitasi inovasi dalam pemuliaan.
Di samping metode tersebut, seseorang dapat mempertimbangkan gambaran genomik yang lebih rinci dengan meningkatkan kepadatan penanda. Secara teori, jumlah penanda yang diperlukan untuk kemampuan prediksi genomik yang tinggi meningkat dengan ukuran populasi efektif dari set data karena ukuran blok tautan menurun (Meuwissen, 2009 ). Namun, manfaat dari peningkatan kepadatan penanda telah ditemukan berkurang melampaui sekitar 5000 penanda (Zhao et al ., 2021 ), menggunakan seri eksperimen 2–4 dari studi ini. Karena ukuran populasi efektif dari studi ini tetap pada tingkat yang sama, tampaknya tidak mungkin bahwa menggunakan lebih dari 13.000 penanda yang digunakan dalam studi ini akan memiliki banyak potensi untuk perbaikan. Faktor pembatas ketika membandingkan ukuran populasi efektif dari studi ini dan studi Zhao et al . ( 2021 ) adalah karena hibrida tidak digunakan untuk prediksi genomik dalam studi ini, jumlah penanda yang digunakan berbeda dan data genomik yang diperhitungkan memainkan peran yang lebih besar dalam studi kami. Pendekatan lain untuk memodelkan korelasi nilai genetik yang lebih baik untuk GBLUP adalah dengan membuat matriks korelasi khusus sifat. Menggunakan matriks hubungan genomik untuk tujuan ini adalah penyederhanaan yang mengasumsikan bahwa lokus efektif jumlahnya sangat besar dan menyebar ke seluruh genom secara merata. Sebaliknya, matriks korelasi genetik dapat dihitung, misalnya, dari hasil Studi Asosiasi Genom-Lebar (GWAS). Untuk menentukan korelasi genetik, hanya penanda yang terkait dengan sifat yang diinginkan yang akan dipertimbangkan, atau dengan menentukan penanda sintetis yang terkait dengan haploblok tertentu (Jiang et al ., 2018 ; Weber et al ., 2023 ).
Selain mempertimbangkan apakah data set pelatihan memiliki kekuatan yang cukup untuk menangkap arsitektur genetik sifat tersebut, seseorang juga harus mempertimbangkan apakah nilai genotipe itu sendiri diukur dengan cukup akurat. Secara khusus, interaksi genotipe-kali-lingkungan dapat membatasi kemampuan prediksi dalam studi ini. Ketika banyak lingkungan diambil sampelnya, proporsi interaksi genotipe-kali-lingkungan dalam varians fenotipik hasil gabah ditemukan besar, terkadang bahkan lebih besar daripada efek genetik aditif (De Los Campos et al ., 2020 ; Jarquín et al ., 2014 ; Lado et al ., 2016 ). Ketika lebih sedikit lingkungan yang diambil sampelnya, dampak interaksi genotipe-kali-lingkungan diperkirakan kurang penting (Montesinos-López et al ., 2024 ). Melanjutkan tren ini, di mana hanya satu lingkungan yang dijadikan sampel, dan dengan demikian desain eksperimen mengacaukan interaksi genotipe-kali-lingkungan dengan efek utama genotipe, GBLUP dapat mencapai akurasi prediksi hasil gabah hingga satu (Norman et al ., 2018 ). Menariknya, lingkungan yang dijadikan sampel dalam jumlah yang lebih besar tidak selalu memerlukan plot yang lebih banyak untuk diuji. Tumpang tindih antara lingkungan juga dapat ditingkatkan dengan melakukan uji hasil gabah pendahuluan menggunakan lebih banyak lingkungan dari jaringan pengujian, mendistribusikan kandidat secara jarang dan dengan demikian menjaga jumlah plot keseluruhan tetap konstan. Hal ini telah terbukti menguntungkan dalam penelitian sebelumnya untuk seri hibrida 2 dan 3 (Lell et al ., 2021 ; Zhao et al ., 2021 ), serta untuk populasi biparental barley dan jagung (Endelman et al ., 2014 ). Dalam uji coba pemuliaan yang tidak seimbang dari studi ini, bagian terbesar dari data terdiri dari varietas kandidat tahap awal yang diuji dalam kurang dari lima lingkungan, dan hampir tidak ada genotipe uji seri dalam lebih dari 10 lingkungan per tahun (Tabel 1 ). Selain itu, jumlah tahun per genotipe yang diuji kecil. Oleh karena itu, orang dapat berharap bahwa interaksi genotipe-kali-lingkungan sebagian tercampur dengan efek utama genetik dalam BLUE lintas lingkungan. Asumsi ini didukung oleh pengamatan kami bahwa mengingat jumlah genotipe pelatihan yang sama, meningkatkan jumlah tahun dalam set pelatihan meningkatkan hasil prediksi (Gambar 7b)). Prediksi lintas seri dalam studi ini menggunakan BLUE dari satu seri dalam set uji. Oleh karena itu, dapat dibayangkan bahwa prediksi genom lintas seri akan menghasilkan estimasi nilai pembiakan yang sebenarnya kurang terpengaruh oleh interaksi genotipe-kali-lingkungan yang membingungkan daripada BLUE seri tunggal yang dibandingkan. Interaksi genotipe-kali-lingkungan yang membingungkan dalam BLUE set uji karenanya akan menurunkan kemampuan prediksi. Jika efek ini terbukti relevan, prediksi lintas seri juga akan bernilai bagi pemulia yang set datanya melebihi ambang batas yang disebutkan sebelumnya dari ukuran set pelatihan sekitar 4000 genotipe. Varietas kandidat diuji dalam serangkaian lingkungan yang luas selama prosedur pendaftaran resmi, yang melebihi jumlah lingkungan tahap pembiakan akhir. Oleh karena itu, pembandingan metode prediksi genom menggunakan genotipe tahap akhir dari program pembiakan sendiri dapat memberikan hasil yang terlalu optimis karena residu yang membingungkan interaksi genotipe–kali–lingkungan. Oleh karena itu, memperoleh data fenotipik dan genomik dari uji varietas resmi akan menjadi standar emas untuk menilai dampak prediksi lintas seri terhadap kemampuan prediksi. Upaya ini mudah terhambat oleh masalah hukum, tetapi tampaknya bermanfaat.
Metode
Bahan tanaman dan uji lapangan
Delapan seri eksperimen berbeda disusun untuk penelitian ini. Empat seri pertama terdiri dari data historis yang telah dipelajari sebelumnya. Selain itu, empat seri lagi disumbangkan oleh perusahaan pemuliaan untuk penelitian ini. Dalam seri eksperimen tersebut, jumlah genotipe berkisar antara 380 hingga 5051 dan jumlah lingkungan (kombinasi lokasi-tahun) berkisar antara 8 hingga 61 (Tabel 1 ). Seri-seri tersebut dicirikan sebagai berikut:
- Seri 1: Uji coba hasil gabah ortogonal dari 380 genotipe yang mewakili keragaman luas kelompok pemuliaan elit Eropa (Gogna et al ., 2022 ).
- Seri 2: Uji coba hasil gabah non-ortogonal dari 1.099 galur murni yang beragam, termasuk galur unggul dan sumber daya genetik tanaman, serta 5.051 hibrida (Zhao et al ., 2021 , Seri eksperimen II hingga V).
- Seri 3: Uji coba hasil gabah ortogonal dari 135 galur induk elit, 1604 hibridanya, dan 10 varietas yang dirilis (Zhao et al ., 2021 , Seri Eksperimen I, dan Zhao et al ., 2015 ).
- Seri 4: Uji coba hasil gabah non-ortogonal, dihasilkan selama program pemuliaan galur murni komersial, terdiri dari 4958 genotipe (Zhao et al ., 2021 , Seri Eksperimen VI).
- Seri 5–8: Empat seri percobaan, disediakan oleh empat perusahaan pemuliaan. Seri tersebut terdiri dari 781–3516 genotipe yang diuji di rata-rata 1,3–6,7 lokasi dalam 2 tahun (Tabel 1 ). Keempat seri tersebut merupakan kutipan dari kegiatan pemuliaan perusahaan yang biasa dilakukan dan oleh karena itu merupakan uji coba hasil gabah non-ortogonal di mana galur-galur diberi fenotipe dan dipilih hingga 3 tahun. Ada tiga sub-uji coba per seri, yang mencerminkan tahap-tahap uji coba pemuliaan. Kandidat dievaluasi untuk hasil gabah di bawah perlakuan intensif dan sebagian untuk tanggal pembungaan dan tinggi tanaman. Konektivitas antara data dari setiap seri dipastikan oleh beberapa genotipe umum (Tabel S1 ).
Analisis data fenotipik
Kami menerapkan pendekatan dua langkah untuk menganalisis data fenotip seperti yang dijelaskan sebelumnya (Lell et al ., 2024 ) termasuk koreksi outlier mengikuti metode M5 dari Bernal-Vasquez et al . ( 2016 ). Lihat Gambar 1 untuk ikhtisar. Penaksir linier tak bias terbaik dari genotipe (BLUE) di dalam dan lintas lingkungan diperoleh setelah koreksi outlier. BLUE dalam lingkungan dihasilkan menggunakan model linier campuran berikut:
![]()
Di mana
adalah data hasil gabah tingkat plot,
adalah rata-rata keseluruhan,
adalah efek genotipe,
Bahasa Indonesia:
Dan
adalah efek desain untuk uji coba, replikasi dan blok,
adalah efek sisa, dan
Bahasa Indonesia:
Bahasa Indonesia:
Dan
adalah indeks untuk efek model. Bergantung pada desain eksperimen, hanya beberapa atau tidak ada efek yang
Bahasa Indonesia:
atau
diperkirakan. Ketika fenotipe dilakukan dalam percobaan yang tidak direplikasi di suatu lingkungan, efek desain diperkirakan dan dikurangi dari pengukuran. Efek desain diperkirakan sebagai efek acak, g diperkirakan sebagai efek acak untuk menghitung BLUP dan memperkirakan varians genetik dan sebagai efek tetap untuk memperoleh BLUE. Semua efek acak didistribusikan secara identik dan independen. Pengulangan diperkirakan sebagai
, Di mana
adalah estimasi varians genotipe,
adalah estimasi varians residual, dan
adalah jumlah rata-rata plot per genotipe. Lingkungan yang memiliki pengulangan <0,3, atau yang menunjukkan korelasi <0,1 terhadap semua lingkungan lain secara rata-rata dibuang.
BLUE lintas lingkungan diperoleh dengan menggunakan model campuran berikut:
![]()
Di mana
adalah rata-rata genotipe dalam lingkungan,
adalah rata-rata keseluruhan,
adalah efek seri eksperimen,
adalah efek genotipe,
adalah lingkungan (lokasi
tahun) dampaknya,
adalah efek sisa, dan
Bahasa Indonesia:
, Dan
adalah indeks untuk efek model. Efek
diperkirakan sebagai efek tetap,
adalah efek acak, dan
ditetapkan untuk BLUE dan acak untuk BLUP. Heritabilitas dalam arti luas diperkirakan sebagai
, Di mana
adalah jumlah rata-rata lingkungan di mana suatu genotipe diukur, dan
Dan
adalah estimasi varians genetik dan residual, masing-masing. Semua perhitungan dilakukan menggunakan R 4.0.4 (Tim Inti R, 2021 ) yang ditautkan ke OpenBLAS 0.2.20 ( https://www.openblas.net ) dan AsReml-R 4.1.0.110 (Gilmour et al ., 2015 ) pada mesin Linux dengan 4 prosesor Intel Xeon CPU E7-4890 v2 (120 inti logis) dan memerlukan sekitar 500 GB RAM.
Analisis data genotipe
Semua seri eksperimen memiliki data SNP yang tersedia untuk berbagai fraksi genotipe (Tabel 2 ). Data SNP dihasilkan menurut proses masing-masing penyedia data dan dengan demikian pilihan penanda SNP yang heterogen tersedia untuk berbagai seri eksperimen (Gambar 2a ). Panggilan SNP difilter secara individual untuk setiap kelompok genotipe menurut ambang batas berikut: Genotipe dibuang jika memiliki lebih dari 30% nilai yang hilang atau lebih dari 10% panggilan heterozigot. Penanda dibuang jika memiliki lebih dari 90% nilai yang hilang atau lebih dari 20% panggilan heterozigot. Karena data SNP dihasilkan oleh penyedia yang berbeda, kami selanjutnya memeriksa dan mengoreksi konsistensi penunjukan untai menggunakan genotipe yang tumpang tindih sebelum mengintegrasikan data di seluruh seri eksperimen. Efisiensi komputasi dari proses ini ditingkatkan dengan memodelkan tumpang tindih susunan SNP sebagai grafik dan menentukan pohon rentang maksimum (Markowski et al ., 2021 ). Posisi penanda diturunkan menggunakan BLAST (Camacho et al ., 2009 ) dan Chinese Spring Reference Sequence v.2.1 (Zhu et al ., 2021 ), menghapus penanda yang urutannya menunjukkan ketidakcocokan dengan urutan referensi atau dipetakan ke lebih dari satu posisi. Dari penanda yang menunjukkan variasi untuk populasi, posisi fisik yang unik ditemukan untuk 29.970 penanda dan data penanda tersebut kemudian diimputasi. Imputasi dilakukan menggunakan BEAGLE 5.2 (Browning et al ., 2018 ) tanpa menggunakan panel referensi untuk informasi pentahapan, dengan ukuran jendela 1000, ukuran tumpang tindih 100, dan 10 iterasi burn-in.
Karena data SNP bersifat heterogen dalam hal kepadatan penanda, terdapat kesenjangan non-acak dalam data SNP, sehingga sejumlah besar penanda hanya tersedia untuk sebagian kecil individu. Untuk menilai akurasi imputasi kesenjangan sistematis tersebut, kami memilih pendekatan masking terblokir: Kami membagi penanda yang tersedia menjadi set kepadatan rendah dan set kepadatan tinggi berdasarkan fraksi genotipe yang memiliki data untuk penanda masing-masing. Penanda yang lebih dari 70% genotipenya memiliki data dikelompokkan ke dalam set kepadatan rendah; semua penanda lainnya membentuk set kepadatan tinggi (set kepadatan rendah: 2582 penanda, set kepadatan tinggi: 13604 penanda). Untuk 10% genotipe acak yang memiliki data untuk lebih dari setengah penanda kepadatan tinggi, data penanda kepadatan tinggi ditutupi. Set data yang dihasilkan diimputasi menggunakan parameter BEAGLE yang sama seperti yang dijelaskan sebelumnya. Untuk setiap penanda, akurasi imputasi dihitung sebagai jumlah panggilan SNP bertopeng yang data imputasinya cocok dengan panggilan asli dibagi dengan jumlah total panggilan SNP bertopeng. Proses ini diulang sebanyak 20 kali. Dalam percobaan tambahan, kami menggunakan pendekatan masking acak untuk meniru panggilan yang hilang seolah-olah terjadi, misalnya, dalam pendekatan genotyping-by-sequencing. Kami menutupi 1% dari semua panggilan penanda secara acak di seluruh set data, mengimputasikannya, dan menghitung akurasi imputasi per penanda seperti yang dijelaskan di atas. Proses ini juga diulang sebanyak 20 kali.
Untuk memvisualisasikan struktur populasi, kami menghitung jarak Rogers (Rogers, 1972 ) menggunakan set data SNP sebelum imputasi (Gambar 2a ). Kami menyaring data untuk tingkat nilai hilang maksimum 80% per penanda dan frekuensi alel minor minimum 0,05. Ini mempertahankan 13.720 penanda dari data awal. Jarak Rogers dihitung untuk setiap pasangan genotipe berdasarkan penanda yang tersedia untuk keduanya. Jarak Rogers kemudian dikenakan analisis koordinat utama menggunakan fungsi R ‘cmdscale’.
Prediksi genomik dalam dan lintas seri eksperimen
Dalam setiap seri percobaan, korespondensi data fenotipik dan genotipik dinilai. Untuk ini, sampel acak 90% dari rata-rata genotip dalam setiap seri diambil sebagai set pelatihan untuk memprediksi 10% sisanya. Proses ini diulang 20 kali untuk setiap seri percobaan. Untuk mendapatkan rata-rata genotip dalam setiap seri, model (2) dipasang pada BLUE dalam lingkungan tanpa efek seri.
Efek genotipe yang dihasilkan (BLUE dalam seri) kemudian digunakan sebagai pengukuran dalam model BLUP genomik (GBLUP):
![]()
di mana y adalah BLUE dalam seri,
adalah rata-rata,
adalah efek sisa, dan
adalah efek genotipe yang dimodelkan untuk dikorelasikan menggunakan matriks hubungan genomik VanRaden ( 2008 )
, Jadi
, dimana variansnya
diperkirakan oleh model. Data SNP yang digunakan untuk menghitung K difilter untuk hanya menyertakan penanda yang memiliki panggilan SNP untuk setidaknya 80% dari genotipe seri tersebut dan sebagai tambahan frekuensi alel minor setidaknya 0,05. Prediksi dilakukan menggunakan paket BGLR R (Pérez dan de los Campos, 2014 ).
Kami membuat banyak set pelatihan berbeda untuk prediksi genomik dengan memilih berbagai kombinasi seri eksperimen sebagai set pelatihan. Dari sejumlah besar kemungkinan kombinasi, kami memilih satu subset yang dioptimalkan secara numerik untuk desain eksperimen D-optimal menggunakan algoritma Fedorov (Wheeler, 2004 ), dengan 135–349 kombinasi seri berbeda sebagai set pelatihan, tergantung pada ukuran set pelatihan maksimum untuk sifat tersebut. Kami kemudian menentukan kemampuan prediksi seri eksperimen yang tidak termasuk dalam masing-masing set pelatihan.
Untuk menguji pengaruh langkah-langkah kontrol kualitas selama integrasi data, kami melakukan prediksi dengan empat versi data SNP yang berbeda, yang memengaruhi matriks hubungan genomik.
: Penanda difilter menggunakan ambang batas liberal dengan maksimal 80% nilai hilang per penanda atau ambang batas ketat dengan maksimal 30% nilai hilang per penanda. Data yang dihasilkan dari ambang batas liberal memiliki 13.692 penanda. Data yang dihasilkan dari ambang batas ketat memiliki 5.913 penanda. Lebih jauh, kami menggunakan data SNP dengan atau tanpa imputasi oleh BEAGLE. Dalam kasus yang tidak diimputasi, celah dalam data penanda diisi oleh rata-rata penanda masing-masing seperti yang dijelaskan di atas. Untuk jarak VanRaden, yang melibatkan pemusatan data SNP, ini setara dengan mendasarkan jarak berpasangan hanya pada penanda yang memiliki informasi pada kedua individu yang terlibat. Matriks kekerabatan yang dihasilkan digunakan sebagai matriks kovariansi untuk BLUP genomik menggunakan model berikut:
![]()
Di mana
Bahasa Indonesia:
Bahasa Indonesia:
Dan
didefinisikan secara analog dengan (3). Seperti untuk validasi silang dalam seri, perhitungan dilakukan menggunakan paket BGLR R.
Selanjutnya, kami fokus pada analisis yang lebih rinci tentang pengaruh seri individual terhadap kemampuan prediksi. Kami mendasarkan hal ini pada prediksi yang dijalankan dengan data SNP yang diperhitungkan dengan ambang batas nilai hilang yang liberal. Untuk setiap set pengujian
(
, Di mana
, jumlah seri dalam penelitian ini), sebuah model
![]()
disesuaikan untuk memperkirakan peningkatan rata-rata kemampuan prediksi dengan ukuran set pelatihan, di mana
adalah kemampuan prediksi dari rangkaian uji prediksi genomik
yang menggunakan rangkaian seri percobaan
sebagai set pelatihan.
adalah jumlah genotipe dalam set pelatihannya,
Dan
adalah parameter empiris, diperkirakan secara terpisah untuk setiap set pengujian, dan
adalah penyimpangan dari prediksi yang dijalankan
dari rata-rata empiris. Sebagian besar prediksi genomik memiliki lebih dari satu seri sebagai set pelatihan, itulah sebabnya
adalah indeks dan
adalah sekumpulan indeks.
Kami mempelajari bagaimana rangkaian eksperimen individu yang ada dalam set pelatihan mempengaruhi kemampuan prediksi dari set pengujian tertentu dengan menyesuaikan model campuran linier pada vektor
penyimpangan dari kecocokan empiris (5) untuk semua set pelatihan dan pengujian. Berikut ini, kami menunjukkan penyimpangan dari satu prediksi genomik sebagai
, diindeks oleh seri eksperimen sebagai set pengujian
dan setnya
indeks seri eksperimen dalam set pelatihan. Berdasarkan hal ini, model linier
![]()
Model ini menguraikan penyimpangan prediksi genomik menjadi rata-rata
dan tiga kelompok efek acak: Efek utama dari set pengujian (
) dan set pelatihan (
) dengan
efek masing-masing, dan efek dari masing-masing kombinasi efek
dari dua seri eksperimen sebagai set pengujian
dan set pelatihan
Untuk masing-masing
Bahasa Indonesia:
Dan
, satu parameter varians diestimasikan. Karena satu run memiliki lebih dari satu set pelatihan, maka istilah
memilih untuk setiap pengukuran
parameter yang relevan. Sisanya dilambangkan sebagai
Model dipasang menggunakan paket BGLR R sebagai Bayesian Ridge Regression (pengaturan model ‘BRR’).
Jaringan saraf konvolusional untuk prediksi genomik
Untuk membandingkan dengan GBLUP, kami menggunakan Jaringan Syaraf Tiruan Konvolusional (CNN) untuk menilai kemampuan prediksi BLUE dalam seri (bagian sebelumnya) dan BLUE lintas seri. Kerangka kerja Python ‘Keras’ digunakan untuk pengembangan model. CNN beroperasi pada urutan status penanda satu dimensi, yang diurutkan berdasarkan posisi pemetaannya dalam genom. Dua status homozigot dikodekan sebagai 0 dan 2, dan status heterozigot sebagai 1. Vektor status fenotipik dan penanda diskalakan ulang ke rentang [0, 1].
Untuk set pelatihan, kami memilih sampel acak sebesar 10%, 30%, 60%, dan 80% dari total data yang tersedia untuk setiap sifat. Set pengujian terdiri dari 100 genotipe yang tidak ada dalam set pelatihan, dipilih secara acak untuk setiap iterasi. Proses ini diulang selama 20 kali pengujian untuk setiap sifat dan ukuran set pelatihan. CNN dirancang untuk memungkinkan arsitektur jaringan yang fleksibel untuk setiap pengujian, dengan neuron sebagai tepi dalam grafik berarah asiklik, yang disusun menjadi beberapa lapisan untuk menangkap struktur hubungan dan haplotipe, dan interaksi genetik, masing-masing.
Rangkaian lapisan pertama difokuskan pada ekstraksi fitur, diikuti oleh lapisan untuk pengenalan pola. Struktur jaringan spesifik dipengaruhi oleh berbagai hiperparameter, yang nilainya dioptimalkan menggunakan Hyperband Tuner (Li et al ., 2017) ). Ruang hiperparameter untuk bagian ekstraksi fitur mencakup (1) jumlah lapisan konvolusi dan pengumpulan rata-rata bergantian (tiga hingga lima), (2) jumlah filter (berkisar dari 64 hingga 512 dengan ukuran langkah 64) dan (3) ukuran kernel (antara tiga dan 36 dengan ukuran langkah tiga) di lapisan konvolusi. Ukuran kumpulan untuk lapisan pengumpulan juga divariasikan antara dua dan 32 dengan ukuran langkah 4. Keluaran ekstraksi fitur diratakan dan diteruskan ke bagian pengenalan pola.
Ruang hiperparameter untuk bagian pengenalan pola terdiri dari (1) jumlah lapisan padat dan lapisan putus yang bergantian (satu hingga empat), (2) jumlah unit lapisan padat (antara 32 dan 256 dengan ukuran langkah 32) dan (3) rasio putus (antara 0,1 dan 0,5 dengan ukuran langkah 0,01). Putus diterapkan untuk mengurangi overfitting dengan mencegah model menjadi terlalu bergantung pada neuron tertentu. Fungsi Rectified Linear Unit (ReLU) digunakan sebagai fungsi aktivasi untuk semua lapisan kecuali neuron prediksi akhir. Neuron tunggal dengan fungsi aktivasi tangen hiperbolik (tanh) melakukan prediksi sifat, menerima masukan dari semua neuron di lapisan pengenalan pola akhir.
Untuk setiap set hiperparameter yang dipilih oleh penyetel, model dipasangkan dengan ukuran batch sebanyak 32 genotipe. Set pelatihan dibagi menjadi 90% yang akan digunakan untuk tujuan ini dan 10% yang digunakan secara eksklusif untuk membandingkan kinerja prediksi model yang dilatih yang dihasilkan dari pilihan hiperparameter (set validasi). Sasaran penyetel adalah untuk meminimalkan kesalahan kuadrat rata-rata antara rata-rata genotipe yang diprediksi dan yang diamati dalam set validasi. Penyetelan hiperparameter dihentikan ketika kesalahan tidak berkurang lebih dari 0,01 selama 5 iterasi atau lebih. Model yang dihasilkan digunakan untuk prediksi genomik.
Sebagai perbandingan dengan GBLUP, data lintas seri yang sama seperti yang digunakan untuk Jaringan Saraf dikenakan GBLUP mengikuti Persamaan ( 4 ).
Pengaruh jumlah seri eksperimen, lingkungan dan tahun pada set pelatihan
Kami mengukur efek dari penyertaan satu seri eksperimen tunggal versus beberapa seri eksperimen ke dalam set pelatihan, pada jumlah genotipe yang konstan sebanyak 800 dalam set pelatihan dan 100 genotipe dalam set pengujian. Kedua set dipilih secara acak (25 replikasi). Dalam skenario pertama, kami memilih semua genotipe pelatihan dari satu seri eksperimen acak saja. Hanya seri eksperimen 6, 7 dan 8 untuk tanggal pembungaan dan tinggi tanaman dan seri 2, 4, 6, 7 dan 8 untuk hasil gabah yang cukup besar untuk ini. Dalam skenario kedua, kami mengambil sampel sejumlah genotipe yang sama secara acak dari semua seri kecuali satu. Set pengujian diambil sampelnya dari seri yang tidak ada dalam set pelatihan.
Kami selanjutnya menyelidiki strategi potensial untuk membuat subset genotipe berdasarkan kualitas data atau jumlah lingkungan fenotipe untuk meningkatkan prediksi yang diperoleh dari set data terintegrasi yang besar dengan membatasi set pelatihan untuk memenuhi kriteria yang berbeda. Untuk menguji pengaruh pada kemampuan prediksi jumlah lingkungan yang menjadi dasar set pelatihan, kami menugaskan semua genotipe ke salah satu dari empat kelompok lingkungan: 1–3, 4–5, 6–9 dan 10 atau lebih lingkungan. Ini dipilih berdasarkan data yang tersedia untuk memiliki kelompok yang ukurannya sama mungkin sambil tetap memasukkan genotipe dari beberapa seri eksperimen ke dalam satu kelompok lingkungan. Set pelatihan terdiri dari 300 genotipe yang dipilih secara acak dari genotipe kelompok lingkungan. Set pengujian adalah 100 genotipe yang diambil secara acak sambil memastikan bahwa genotipe dari setiap seri eksperimen disertakan dalam bagian yang sama dalam set pengujian. Untuk setiap kelompok lingkungan, 25 replikasi prediksi dilakukan. Untuk prediksi, model GBLUP (3) digunakan, dan matriks kekerabatan didasarkan pada data SNP yang diimputasikan menggunakan kriteria nilai hilang liberal (13.692 penanda).
Untuk menguji apakah data historis masih berguna untuk prediksi, kami memilih set pelatihan yang memiliki jumlah genotipe yang sama tetapi berbeda dalam jumlah tahun yang dicakup oleh data pelatihan. Kami memilih rentang 2 tahun dari mana kami mengambil sampel 600 genotipe pelatihan untuk setiap rentang: 1–2 tahun sebelum set pengujian dan 1–5 tahun sebelum tahun set pengujian. Data kami mencakup 12 tahun yang berbeda, sehingga kami dapat menghasilkan set pengujian selama 7 tahun (tahun ke-6 hingga tahun ke-12). Analisis hanya dilakukan untuk hasil gabah karena data yang tersedia untuk tanggal pembungaan dan tinggi tanaman tidak ada untuk beberapa lingkungan. Kami juga mengecualikan satu set pengujian (2020) karena tahun-tahun sebelumnya tidak memungkinkan untuk set pelatihan dengan ukuran yang memadai. Set pengujian mencakup 100 genotipe acak yang diukur pada tahun masing-masing. Seperti yang disebutkan sebelumnya, kami menggunakan kumpulan BLUE lintas lingkungan yang telah dihitung sebelumnya sebagai data untuk prediksi, jadi secara teknis informasi dari lebih dari 1 tahun disertakan dalam beberapa BLUE ini. Namun, sebagian besar genotipe dalam data kami diukur hanya dalam 1 tahun. Kami memastikan bahwa genotipe yang disertakan dalam set pelatihan tidak disertakan dalam set pengujian. Prediksi dilakukan menggunakan model GBLUP (3) dan matriks kekerabatan didasarkan pada data SNP besar yang diperhitungkan.


Leave a Reply